The Protein Folding Problem
Why shape is life’s code

Protein Folding
At the very core of virtually every process in a living organism are proteins. Think of them as microscopic machines, each meticulously designed for a specific job. They are the workhorses of our cells, responsible for everything from digesting our food and fighting off infections to providing structural support and transmitting signals in our brains.
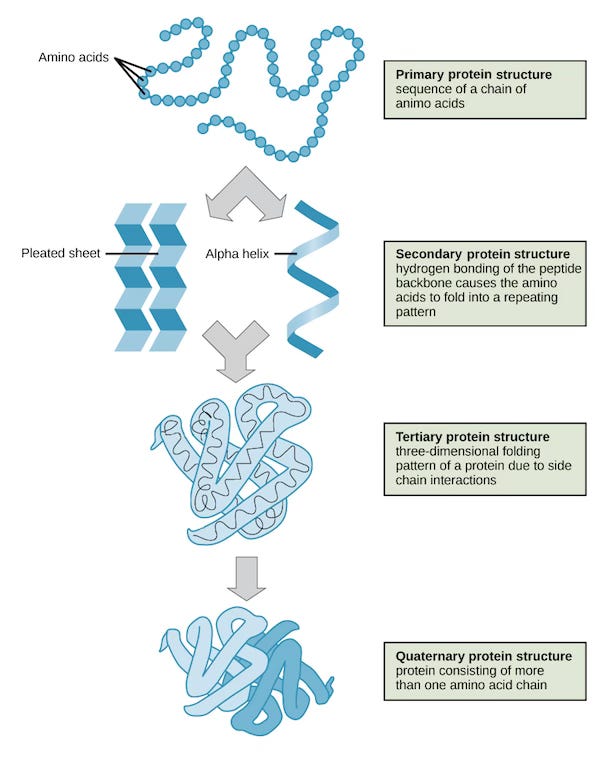
Proteins begin as a simple, linear chain of building blocks called amino acids. This initial sequence is known as the primary structure. However, this chain doesn't stay linear for long. Almost instantaneously, it begins to twist and fold into a complex and highly specific 3D structure. This folding process is governed by the chemical interactions between the amino acids in the chain.
This intricate dance of folding gives rise to:
Secondary Structure: Localized folding patterns like alpha-helices (corkscrews) and beta-sheets (pleated sheets).
Tertiary Structure: The overall 3D shape of a single polypeptide chain, bringing together different secondary structures. This is the functional form for many proteins.
Quaternary Structure: The arrangement of multiple polypeptide chains (subunits) to form a larger, functional protein complex.
The following image is taken from Khan Academy.
Why the Shape of a Protein Matters
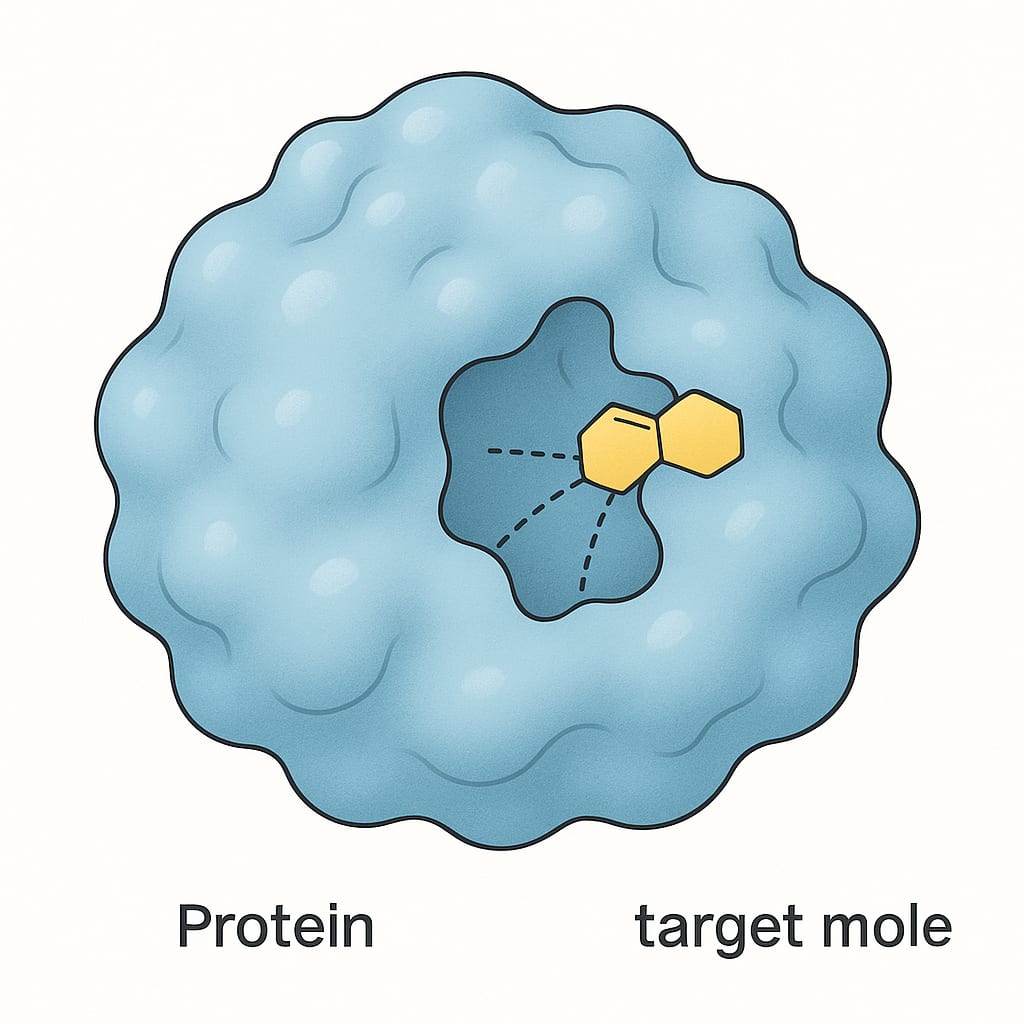
The principle "structure dictates function" is a cornerstone of biology. The specific 3D shape of a protein is what allows it to perform its designated role. For instance, the pocket-like shape of an enzyme's active site is perfectly molded to bind to its specific target molecule, much like a key fits into a lock or lego blocks. Similarly, the shape of an antibody protein allows it to recognize and bind to specific invaders like viruses and bacteria.

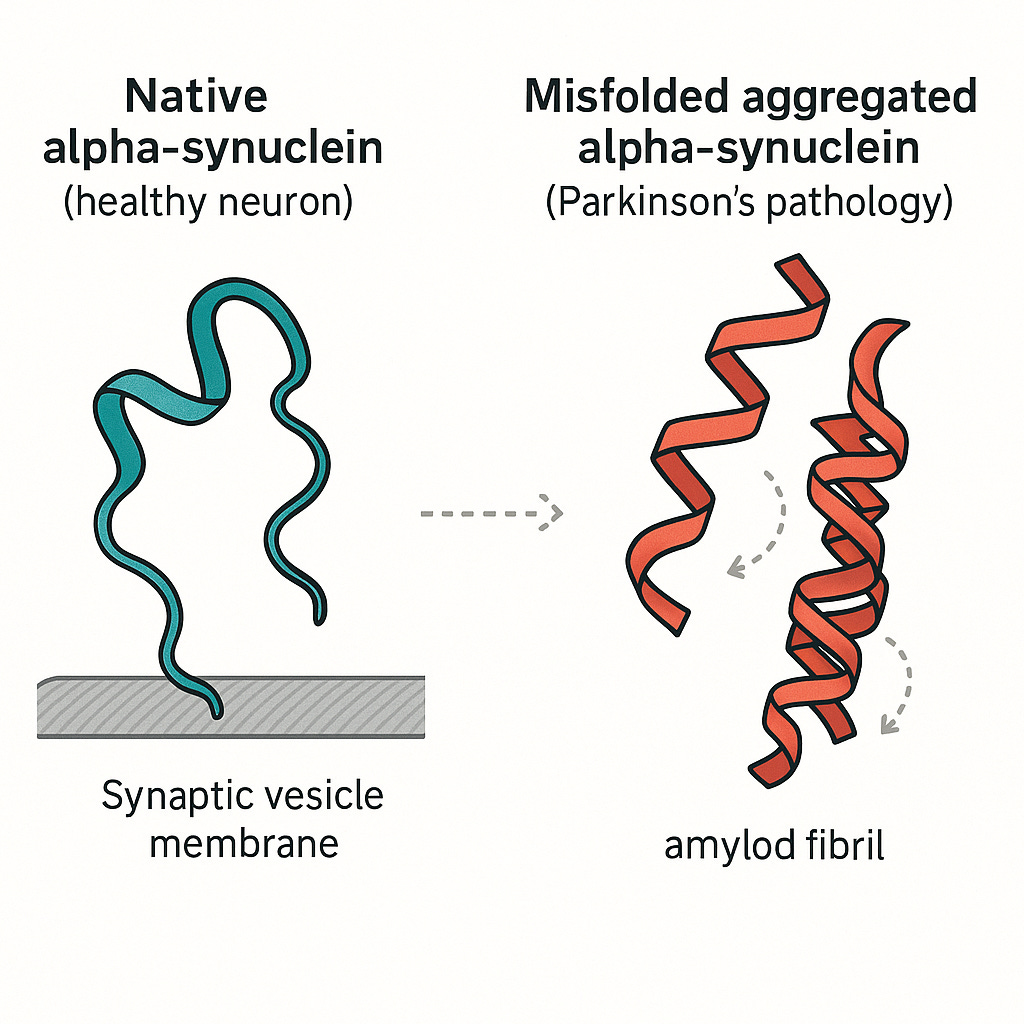
When proteins fold correctly, our bodies function as they should. However, when they misfold, the consequences can be devastating. Misfolded proteins can lose their function or, even worse, clump together and become toxic to cells. This process is now understood to be a key driver of numerous debilitating diseases, including Alzheimer's Disease, Parkinson's Disease, Cystic Fibrosis etc.
By understanding a protein's structure, scientists can gain invaluable insights into its function in both health and disease. This knowledge is paramount for designing new drugs that can target specific proteins, correcting the effects of misfolding, and developing novel therapies for a wide range of medical conditions.
🔑 The Key Idea: A protein's function depends entirely on its 3D shape. It's like a key: its specific pattern of ridges and grooves is what allows it to open a particular lock. If the key is misshapen, the lock won't turn.
Protein Folding Problem
Given that a protein’s function is determined completely by its 3D folding, how do we understand the shape of a protein.
A fundamental principle was first demonstrated in the 1961 by Christian Anfinsen. In a landmark experiment, he took a protein, ribonuclease-A, and used harsh chemicals to force it to unroll into a useless, floppy thread. When he removed the chemicals, something amazing happened: the protein spontaneously folded back into its exact original shape and started working again. This proved that all the information needed to create a protein's complex 3D structure is encoded directly within its amino acid sequence. This concept, known as Anfinsen's thermodynamic hypothesis, laid the groundwork for the entire field. It gave rise to the "protein folding problem" which means predicting the final, functional 3D structure from its primary amino acid sequence alone.
If structure emerges so easily, why is it hard to predict? In 1969 Cyrus Levinthal estimated that a 100-amino acid protein has ~10¹⁰⁰ possible conformations. Exhaustively sampling them would take longer than the universe has existed, yet real proteins fold almost instantly. Levinthal’s paradox hinted at an energy-landscape “funnel” guiding the chain toward its native state - and at the computational nightmare of mapping that landscape. The protein folding problem is like trying to predict the final, complex shape of an origami creation by only knowing the sequence of folds.
For decades, determining the 3D structure of a protein was a monumental task. Through most of the twentieth century structural biology relied on X-ray crystallography, nuclear-magnetic-resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM). Each has strengths, but all are slow and technically demanding. The impact of cryo-EM was dramatic enough to earn Jacques Dubochet, Joachim Frank and Richard Henderson the 2017 Nobel Prize in Chemistry. Furthermore, traditional computational methods struggle to accurately predict protein structures from their amino acid sequences due to the sheer complexity and the astronomical number of possible conformations a protein could theoretically adopt.
At the root of it, the computational problem is fairly simple to state:
Design a function that takes a sequence of amino acids as input and returns the 3D coordinates of the amino-acids.The Critical Assessment of Structure Prediction (CASP) is a biennial, blind experiment that has functioned as protein-folding research’s impartial scorecard since its inauguration in 1994. In each round, organisers quietly select newly solved but still-embargoed experimental structures and release only their amino-acid sequences; modelling groups worldwide have a few weeks to submit 3-D predictions, which independent assessors later compare to the unreleased crystal, NMR or cryo-EM coordinates. This format strips away any hint of hindsight, so CASP cleanly tracks genuine methodological progress.
Why machine learning might be effective at the protein folding puzzle
The protein folding problem, at its heart, is a prediction problem - something which machine learning has seen success. What makes it particularly challenging is deciphering an incredibly complex set of patterns. Machine learning models, particularly deep neural networks, are exceptionally good at learning such intricate patterns from vast amounts of data. Here's why ML is the perfect tool for the job:
Vast Data Availability: Over the years, researchers have experimentally determined the structures of hundreds of thousands of proteins and deposited them in public databases like the Protein Data Bank (PDB). This wealth of data provides the perfect training ground for ML models.
Learning from Patterns: An ML model can be trained on these known protein structures. By analyzing the amino acid sequences and their corresponding final shapes, the model learns the subtle and complex relationships between them. It learns how different amino acid combinations and their positions in the sequence influence the final fold.
Predictive Power: Once trained, the model can be given a new amino acid sequence it has never seen before and predict its 3D structure with remarkable accuracy.
We can train a ML model on available data i.e., pairs of amino acid sequences and 3D coordinates for experimented verified proteins. Since the output of the model will be (N,3) matrix where N is the number of amino acids in the sequence, you can use root mean square distance (RMSD) between the actual and predicted coordinates as a loss function. Most advanced models use more sophisticated loss functions e.g. that are invariant to rotations and translations.
AlphaFold-2
AlphaFold-2 is a deep learning model that introduced a novel architecture integrating evolutionary and physical information about proteins. By leveraging deep learning techniques, AlphaFold has been able to predict protein structures with an accuracy that rivals experimental methods. The key insights in AlphaFold-2 system are:
Use sequences of related proteins (homologs) i.e. proteins with common ancestry for every input sequence to provide evolutionary hints to the model.
A novel transformer-based module (Evoformer) to learn co-evolutionary patterns and geometric constraints simultaneously.
A lightweight, equivariant structure module then converts learned features into atomic coordinates, and by recycling its own predictions through the network several times it progressively refines its output.
This showed that you can teach a neural network to “read” the evolutionary history of a protein and turn that into a precise 3D shape, without hand-tuned energy functions or physics simulations.
AlphaFold-2 has been able to predict protein structures with an accuracy that rivals experimental methods, transforming protein structure prediction from a slow, laborious lab task into an almost entirely AI-driven process. We now have highly accurate predictions for nearly every known protein - over 200 million structures have been released publicly. These predictions are often on par with experimentally determined structures. This leap forward is poised to accelerate biological research and drug discovery at an unprecedented rate. Scientists can now rapidly understand disease-causing mutations, design novel drugs with incredible precision, and even engineer new enzymes for industrial applications. By solving the protein folding problem, AlphaFold has not just answered a long-standing scientific question, it has unleashed a new era of biological discovery.
ColabFold
You can play with AlphaFold-2 predictions in a Colab notebook ColabFold which for a given protein sequence, searches its homologs over a database, and then runs the AlphaFold-2 model to predict the 3D structure.
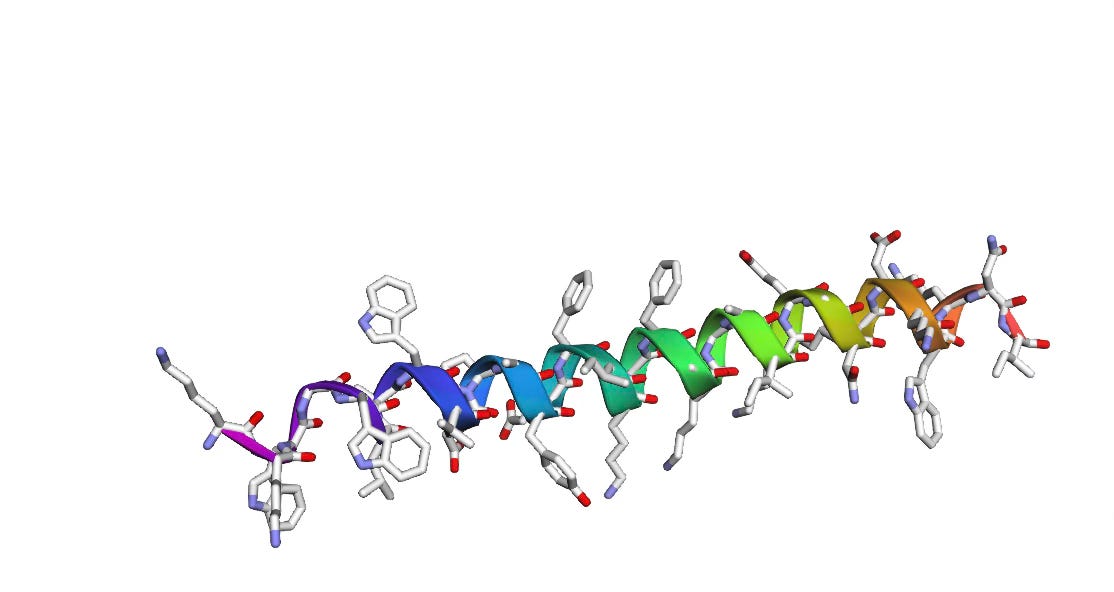
For the amino acid sequence: KKWGWLAWVDPAYEFIKGFGKGAIKEGNKDKWKNI, we get the following 3D structure as predicted by AlphaFold-2
Beyond AlphaFold-2
Academic labs and industry rushed to democratise or extend the breakthrough. Baker Lab’s RoseTTAFold appeared in 2021, offering near-AlphaFold accuracy with open-source code. Noteably, David Baker is the half-recipient of the 2024 Nobel Prize in Chemistry with other half to AlphaFold-2 creators John Jumper and Demis Hassabis of Deepmind. Meta AI’s ESMFold followed, showing that large protein-language models can skip the expensive multiple-sequence alignment step entirely.
And the frontier keeps moving: 2024 saw the debut of AlphaFold 3, incorporating diffusion models to predict not just proteins but their complexes with DNA, RNA and small molecules - a leap toward modeling full cellular machinery.
Journey Continues
This post launches my new AI × Biology series. Over the coming instalments we’ll roll up our sleeves and actually train neural networks to predict protein structures - starting with toy datasets and simple architectures, then layering in multiple-sequence alignments, geometric losses and attention blocks until we inch toward a mini-AlphaFold model. Along the way I’ll zoom out to explore other crossroads of machine learning and life sciences: generative protein design, small-molecule docking, single-cell omics, and whatever breakthroughs surface next. I hope you’ll join me for the journey.
References
AlphaFold paper: Highly accurate protein structure prediction for the human proteome
If you find this post useful, I would appreciate if you cite it as:
@misc{verma2025protein-folding-problem,
title={The Protein Folding Problem: Why shape is life’s code
year={2025},
url={\url{https://januverma.substack.com/p/the-protein-folding-problem}},
note={Incomplete Distillation}
}